Genetic Engineering Attribution¶
Synthetic biology offers fantastic benefits for society, but its anonymity opens the door for reckless or malicious actors to cause serious harm. Thanks to modern machine-learning techniques, reliable attribution of real-world engineered sequences is now within reach – as demonstrated by the results of this competition.
Will Bradshaw, Competition Director at altLabs
Genetic engineering is the process of modifying an organism's DNA to alter its function. Its use has become increasingly widespread, accessible, and powerful. The benefits of this progress is undeniable, but the tools to ensure transparency and accountability are lagging. Currently, there is no easy way of tracing genetically engineered DNA back to its lab-of-origin. This task is known as attribution, and it's a pivotal part of ensuring that genetic engineering progresses responsibly.
When manipulating DNA, a designer has many decisions she must make, such as promoter choice and cloning method. These choices leave clues in the genetic material, and together, compose a "genetic fingerprint" that can be traced back to the designer.
The goal of the Genetic Engineering Attribution Challenge was to develop tools that help human decision makers identify the lab-of-origin from genetically engineered DNA. This competition was composed of two tracks. In the Prediction Track, participants competed to attribute DNA samples to its lab-of-origin with the highest possible accuracy. In the Innovation Track, competitors that beat the BLAST benchmark were invited to submit reports demonstrating how their lab-of-origin prediction models excel in domains beyond raw accuracy.
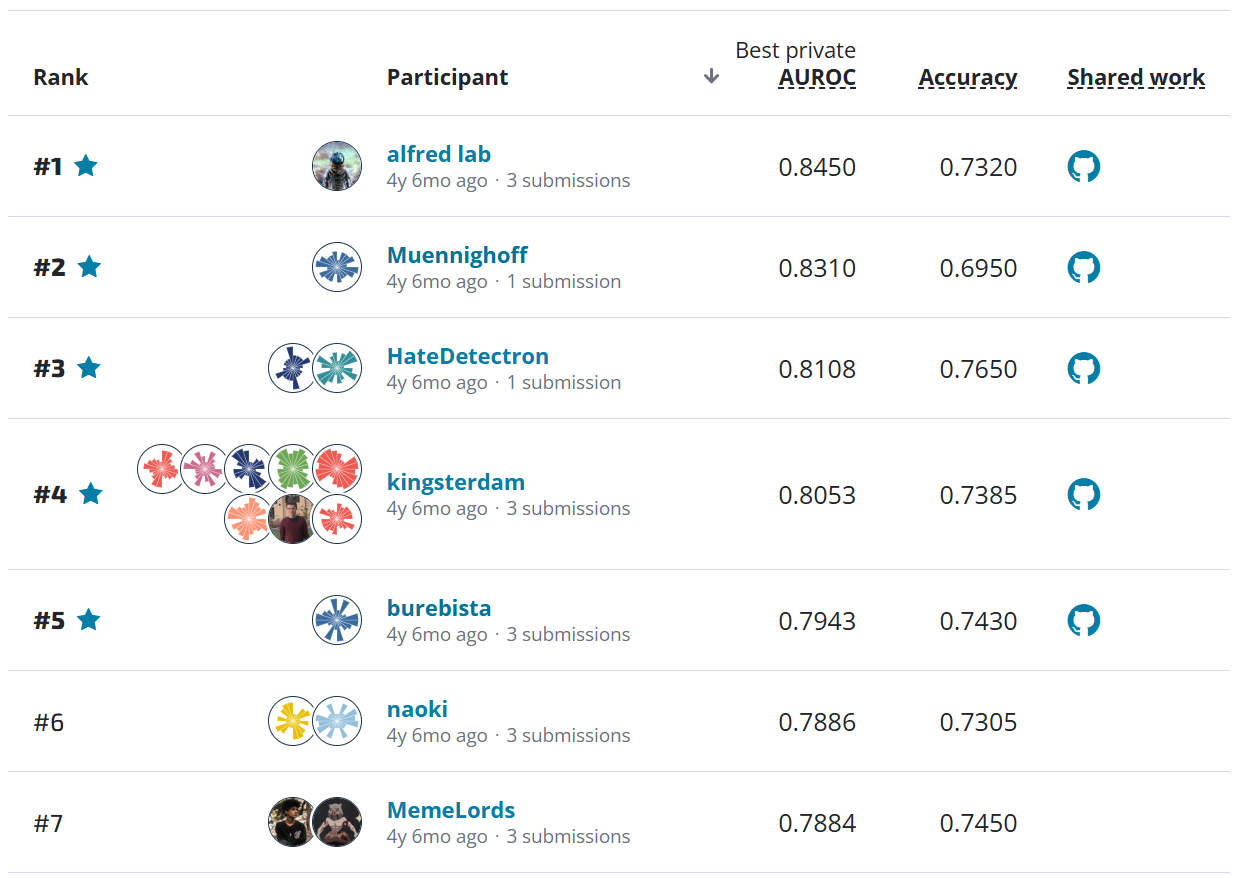
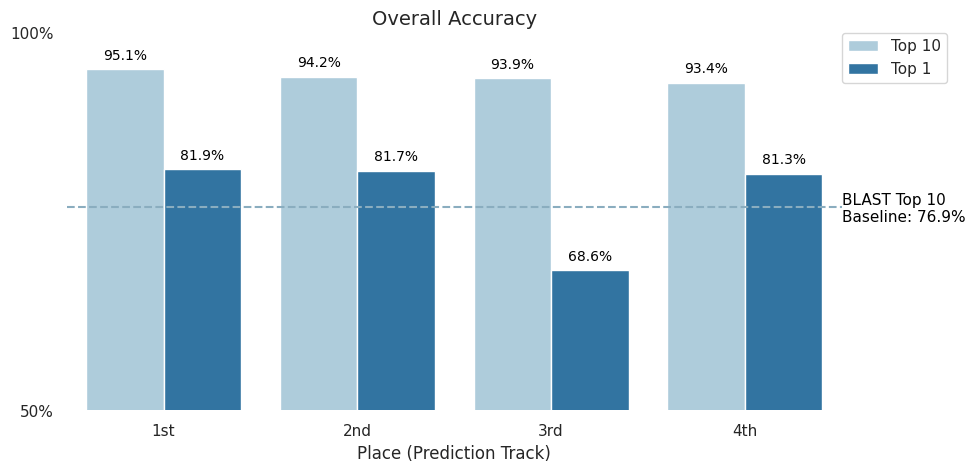
When attributing DNA to its source, simply narrowing the field of possible labs would be a boon to human decision makers. This is why top-10 accuracy was chosen as the competition metric. In the figure below, you can see that the top four Prediction Track submissions were able to beat the BLAST baseline of 76.9% and achieve over 93% top-10 accuracy, with 1,314 possible labs present in the dataset (read more about the data on the competition's Problem Description page). Moreover, three of the top four submissions were able to to achieve more than 80% top-1 accuracy.
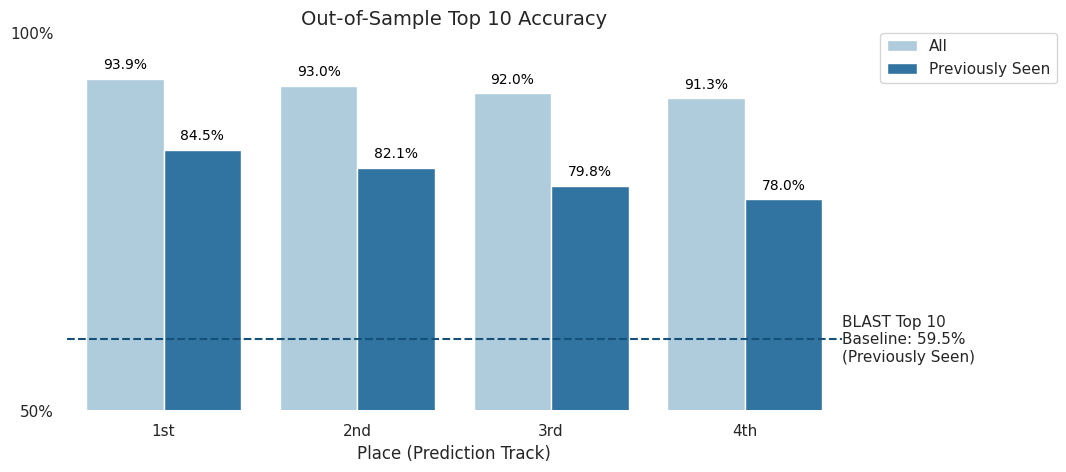
In this competition, solutions were also tested against an out-of-sample verification set from data that was collected after the competition concluded. This test was to ensure that models could perform comparably on unseen data.
The verification data included both "previously seen" labs, or labs that were included in the competition dataset, and "unseen" labs. Predictions for "unseen" labs are considered correct if labeled "unknown".
On the "previously seen" dataset, top competitors in the Prediction Track were able to score an impressive top-10 accuracy of 78% or higher.
To further promote the development and use of attribution technology in the future, altLabs has published all code and models from the winning submissions on Zenodo. In addition, the winners have each provided descriptions of their solutions. Meet the winners and learn how they built their leaderboard-topping attribution models!
Meet the winners¶
Kirill Batmanov¶

Place: 1st Place Prediction and 1st Place Innovation
Prize: $30,000
Hometown: Philadelphia, USA
Username: sorrge
Background:
I am a computational biologist and machine learning enthusiast.
What motivated you to compete in this challenge?
The challenge was about DNA sequence analysis, which is my main area of expertise. However, I never heard about the attribution problem before. It was interesting to see how far I can push the current methods.
Summary of approach:
I made two kinds of models. The first one is based on k-mer counts, because I know from experience that simple k-mer approaches are powerful for DNA sequence matching and comparison. The second is a convolutional neural network with capacity as large as I could make.
The two kinds of models complement each other's strengths well: k-mers are good at precise sequence matching, and work well with low number of samples. CNNs are good at fuzzy motif finding and integrating weak evidence from multiple sites in the sequence. Among the 7 CNNs in the ensemble, 5 are trained directly to predict the lab index, and 2 use self-supervised pre-training of the main CNN layer. The self-supervised target is to predict whether two sequences come from the same lab, using NT-Xent loss.
In the minibatch generation with data augmentation for the CNNs, I use 5% base pair dropout and reverse complement. A random subsequence of 8,000 or 16,000 base pairs is taken. If the original sequence is shorter, it is zero-padded. This setup was discovered by trial and error, and deviation from it lowers the score. In particular, the long (8,000) subsequence length is important.
I use a basic CNN model configuration with a kernel width of 18. It concatenates the sequence representation after max- and average-pooling with the processed binary features. Then a 2-layer MLP makes the final decision. Binary features give a minor boost in accuracy, but they are needed to be competitive.
Roman Solovyev and Ivan Bragin¶

|

|
Place: 2nd Place Prediction and 4th Place Innovation
Prize: $10,000
Hometowns: Moscow, Russia and Voronezh, Russia
Team: fit_dna
Background:
Roman: My name is Roman Solovyev. I live in Russia, Moscow. I’m Ph.D in the microelectronics field. Currently I’m working in the Institute of Designing Problems in Microelectronics (part of Russian Academy of Sciences) as a Leading Researcher. I often take part in machine learning competitions. I have extensive experience with GBM, Neural Nets and Deep Learning as well as with development of CAD programs in different programming languages (C/C++, Python, TCL/TK, PHP etc).
Ivan: My name is Ivan Bragin. I live in Voronezh, Russia. I have a lot of experience in developing high-load services using technologies such as Hadoop, Spark, Cassandra, Kafka. Three years ago, I changed my specialization to machine learning. For two years, I worked as a developer of computer vision algorithms for embedded devices. Currently I'm developing a recommendation system in the Russian social network.
What motivated you to compete in this challenge?
Roman: The problem looked very interesting. I also wanted to play with some Conv1D neural nets.
Ivan: Clear input data and simple baseline. When I reviewed it I already had a lot of ideas and could not stop.
Summary of approach:
The metadata is valuable information but we did not find any insights there. The sequences are even more valuable and after analysis we found some interesting things.
Two DNA sequences generated from the same lab-of-origin often contain similar subsequences. These matched subsequences are the main features that can be extracted from DNA sequences. We decided to search for important subsequences instead of analyzing a sequence as a sentence. This insight helped us to follow the true way and build efficient architecture.
Some provided sequences are read 5′→3′ direction but others are read 3′→5′ direction. This insight was detected based on BLAST output and helped us to improve the quality of models.
Our main idea was to find popular subsequences which describe a lab-of-origin. We tested two approaches:
- Use DNN for automatic sequence analysis: We built a DNN (deep neural network) architecture which automatically extracts subsequences and classifies them. The architecture is presented in the figure below.
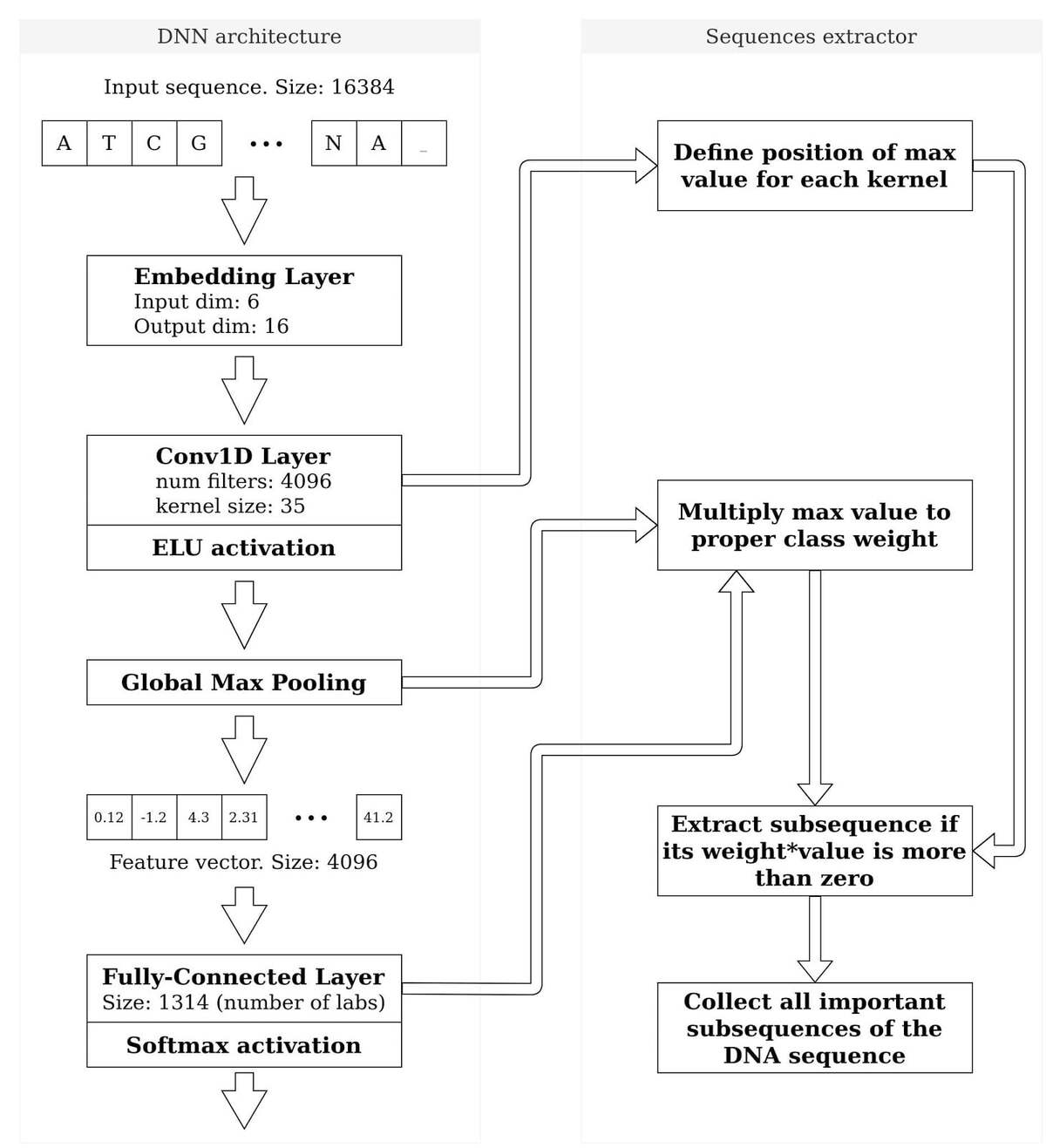
End-to-end DNN model which gets DNA sequence “as is” and returns the vector of lab-of-origin probabilities
- Based on BLAST output: If BLAST finds a similar subsequence for any pair of DNA sequences, we cut it and add it to a set of interesting subsequences. Then we build a matrix of test+train size by number of sequences, about 80,000 by 10^7. This is too big for training. After filtering only popular subsequences and applying Principal Component Analysis (PCA), we extract 1,024 main components and successfully use them to build the lab-of-origin classifier. There are issues in this approach. BLAST doesn’t find all matches in a sequence pair, it reports only the longest one. Furthermore, analysis of 10^7 subsequences requires a lot of hardware resources so we had to reduce it significantly, and thus lost a lot of information.
Evgeny Kuznetsov¶

Place: 3rd Place Prediction
Prize: $5,000
Hometown: Moscow, Russia
Username: eakmail
Background:
Algotrader.
What motivated you to compete in this challenge?
I developed my version of decision trees and wanted to check it in a big competition.
Summary of approach:
I used various n-gram (k-mer in biology) kernels, naive Bayes, soft masks, and rank model merging. I mostly go for fast solutions. I got to the top 7 on my first day with some one-hour n-gram solution. Later I tweaked the kernel to be +4% better and reduced the time down to 4 minutes.
I tried my decision trees library for merging. It still worked better than XGBoost, but simple rank merging worked even better. I also tried advanced n-gram weight formulas.
I used my custom C# research library. It turns C# into something like Python in terms of research speed. Fun fact: I’m probably the first and only C# user prize winner of big ML competitions.
Yao Xie and Grigorii Karmatskii¶

|

|
Place: 4th Place Prediction
Prize: $2,500
Hometowns: Toronto, Canada and St. Petersburg, Russia
Team: what_a_team
Background:
Yao: I am a machine learning enthusiast without formal education in machine learning or computer science. I work as an early childhood educator professionally.
Grigorii: Beginner data scientist.
What motivated you to compete in this challenge?
Yao: The simple data structure and interesting problem setup.
Grigorii: Great way to get familiar with bioinformatics and latest approaches in the field. Opportunity to win prizes. Nice addition to my CV.
Summary of approach:
Yao: I used 100% CNN models since I have been learning deep learning recently. I tried different k-mers but found that 8-mers worked the best. I modeled my solution after this paper: Convolutional Neural Networks for Sentence Classification
Grigorii: I‘m using a simple MLP model, trained on k-mer counts and phenotypic features. It shows high accuracy scores, is quick to train, and doesn’t require a high-end PC.
For feature generation, I counted all 6-mers after extending sequences with its reverse complement. Adding the reverse complement improved quality a lot, and the number of features was reduced after dropping duplicate columns. I also scored using average accuracy per class, which helps to score rare classes.
Adriano Marques, Fernando Camargo, and Igor Muniz¶

|

|

|
Place: 2nd Place Innovation
Prize: $7,500
Hometowns: Chicago, USA and Goiania, Brazil
Team: amalgam
Usernames: adriano_xnv, fernandocamargo, IgorMuniz
Background:
Amalgam: Amalgam is one of the few companies in the market today that is capable of offering a complete suite of solutions for all Artificial Intelligence needs from Server, to Data Center, to Data Engineering, to Data Science, to Machine Learning, and beyond. Our goal is to make Artificial Intelligence easy, cheap, and accessible to companies of all sizes and industries.
Our solutions offer your business the edge required to harness the power of AI to disrupt your industry at a cost effective manner. Your industry is waiting to be disrupted by AI and with Amalgam you can achieve that in a cost-effective manner. Visit us at http://www.amalgam.ai to learn more about how Amalgam can help your business harvest the full power of Artificial Intelligence.
Adriano Marques: Adriano was the engineering team lead at crowdSPRING and is the founder of the Umit Project Open Source Organization where he had, for 6 years, mentored dozens of engineers around the world through the Google Summer of Code program. He is the former co-founder and CTO at Startup Foundry, and currently the CEO of Exponential Ventures, a dream company aimed at creating and launching new Startups and Senior Data Scientist at Loevy & Loevy. A dedicated husband and daddy to two boys and two girls, Adriano still finds time to cook, watch movies, send a camera to the edge of space and record a major storm forming from above, work as a volunteer and read (or listen to) books while he’s not sleeping or working. Since 2012 he's been happily living in the United States with his Family, and in 2016 graduated as a M.Sc. in Artificial Intelligence from Georgia Tech.
Fernando Camargo: Fernando Camargo is a creative, data-driven, and well-rounded Machine Learning Engineer with in-depth machine learning expertise and a wide-ranging 10+ years of experience in software engineering. He is currently a Data Scientist at Exponential Ventures, Machine Learning Researcher at Deep Learning Brazil (Federal University of Goiás), and Co-Founder at DataLIFE.ai. He holds a Masters Degree and is currently pursuing his Ph.D. at Federal University of Goiás.
Igor Muniz: Igor Muniz is a Data Scientist specialized in Machine Learning Engineering. Passionate about puzzles and a competitive person, he has a hobby competing in data science challenges while learning new things every day. He is currently working as Data Scientist at Exponential Ventures, teaching deep learning class and playing chess in his free time.
What motivated you to compete in this challenge?
Everybody in this world has a passion that keeps them moving forward. Our passion is solving the most difficult problems using Artificial Intelligence that can benefit humanity in significant ways. This challenge checked all the boxes. We’re very excited to have placed 10th in the leaderboard and 2nd in the innovation track.
Summary of approach:
Instead of doing a common multi-class classification with a softmax layer, we used Triplet Networks in order to learn embeddings for the labs and learn an embedding extractor for DNA sequences. In this way, we can use distance metrics to rank the labs-of-origin while having the embeddings as a useful sub-product.
The dataset gives us the DNA sequences, some extra inputs, and the lab-of-origin for each of these sequences. We use the DNA sequences and extra inputs to extract an embedding for each sequence. We also have an embedding layer for the labs. We use the lab we get from the dataset as our positive example. Then we do hard negative mining to generate the negative examples. In this case, the negative example is a lab that is the closest to the DNA sequence but is not the lab-of-origin.
We use random roll (rolling the sequence circularly) and cut the DNA to a fixed size before passing it through our model. This way, the model learns to deal with different pieces of the same DNA sequence.
To evaluate, we used top-1 accuracy, top-10 accuracy, and triplet accuracy - the number of times in which the lab-of-origin was the most similar to the DNA sequence divided by the number of examples.
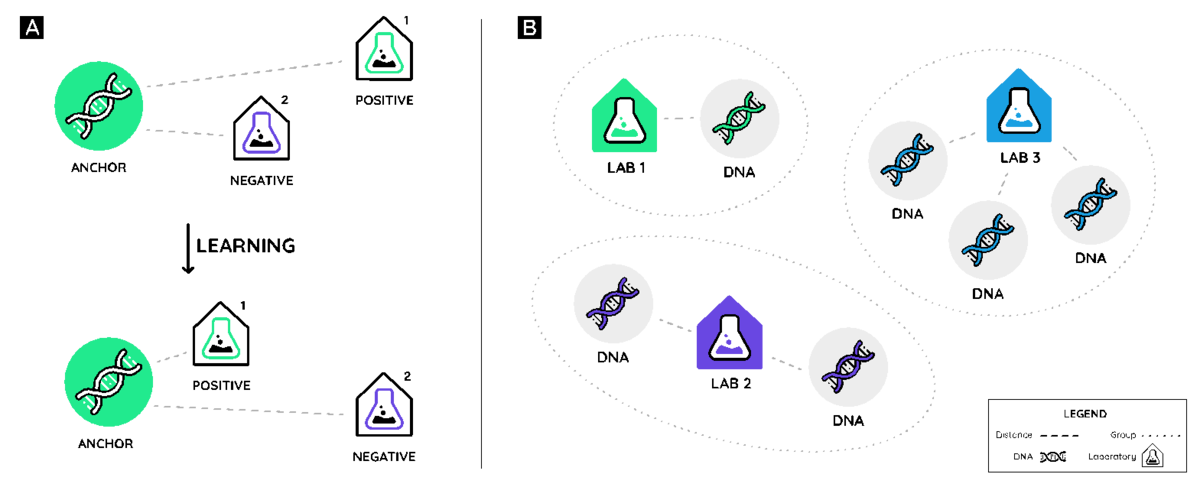
In part A, we learn an embedding such that DNA sequences are closest to their lab-of-origin (positive example), and farther from negative examples. In part B, we use the embeddings to rank the most likely labs for each sequence using distance metrics.
Igor Jovanovic¶
Place: 3rd Place Innovation
Prize: $5,000
Hometown: Serbia
Username: neurogenesis
Background:
I hold a BS in Mechanical Engineering, and PhD in Mathematics. I often like competing on challenge platforms like Herox, Innocentive, and now DrivenData.
What motivated you to compete in this challenge?
I like the sense of having competition, I like winning awards and I enjoy solving challenges.
Summary of approach:
I moved the problem to another dimension by transforming 1D sequences into a 2D image and exploited new relations (statistics, feature extraction, etc.).
Thanks to all the participants and to our winners! Special thanks to altLabs for enabling this important and interesting challenge, and for providing the data to make it possible!